Schrödinger equation
From Wikipedia, the free encyclopedia.
In physics, the Schrödinger equation, proposed by the Austrian physicist Erwin Schrödinger in 1925, describes the time-dependence of quantum mechanical systems. It is of central importance to the theory of quantum mechanics, playing a role analogous to Newton's second law in classical mechanics.
In the mathematical formulation of quantum mechanics, each system is associated with a complex Hilbert space such that each instantaneous state of the system is described by a unit vector in that space. This state vector encodes the probabilities for the outcomes of all possible measurements applied to the system. As the state of a system generally changes over time, the state vector is a function of time. The Schrödinger equation provides a quantitative description of the rate of change of the state vector.
Using Dirac's bra-ket notation, we denote that instantaneous state vector at time t by |ψ(t)〉. The Schrödinger equation is:

where i is the unit imaginary number, 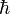 is Planck's constant divided by 2π, and the Hamiltonian H(t) is a self-adjoint operator acting on the state space. The Hamiltonian describes the total energy of the system. As with the force occurring in Newton's second law, its exact form is not provided by the Schrödinger equation, and must be independently determined based on the physical properties of the system.
is Planck's constant divided by 2π, and the Hamiltonian H(t) is a self-adjoint operator acting on the state space. The Hamiltonian describes the total energy of the system. As with the force occurring in Newton's second law, its exact form is not provided by the Schrödinger equation, and must be independently determined based on the physical properties of the system.
Contents |
Time-independent Schrödinger equation
For every time-independent Hamiltonian H, there exist a set of quantum states, |ψn>, known as energy eigenstates, and corresponding real numbers En satisfying the eigenvalue equation

Such a state possesses a definite total energy, whose value En is the eigenvalue of the state vector with the Hamiltonian. This eigenvalue equation is referred to as the time-independent Schrödinger equation. Self-adjoint operators such as the Hamiltonian have the property that their eigenvalues are always real numbers, as we would expect since the energy is a physically observable quantity.
On inserting the time-independent Schrödinger equation into the full Schrödinger equation, we get

It is easy to solve this equation. One finds that the state vectors of the energy eigenstates change by only a complex phase:
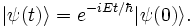
Energy eigenstates are convenient to work with because their time-dependence is so simple; that is why the time-independent Schrödinger equation is so useful. We can always choose a set of instantaneous energy eigenstates whose state vectors {|n>} form a basis for the state space. Then any state vector |ψ(t)> can be written as a linear superposition of energy eigenstates:

(The last equation enforces the requirement that |ψ(t)>, like all state vectors, must be a unit vector.) Applying the Schrödinger equation to each side of the first equation, and using the fact that the energy basis vectors are by definition linearly independent, we readily obtain

Therefore, if we know the decomposition of |ψ(t)> into the energy basis at time t = 0, its value at any subsequent time is given simply by

Schrödinger wave equation
The state space of certain quantum systems can be spanned with a position basis. In this situation, the Schrödinger equation may be conveniently reformulated as a partial differential equation for a wavefunction, a complex scalar field that depends on position as well as time. This form of the Schrödinger equation is referred to as the Schrödinger wave equation.
Elements of the position basis are called position eigenstates. We will consider only a single-particle system, for which each position eigenstate may be denoted by |r>, where the label r is a real vector. This is to be interpreted as a state in which the particle is localized at position r. In this case, the state space is the space of all square-integrable complex functions.
The wavefunction
We define the wavefunction as the projection of the state vector |ψ(t)> onto the position basis:
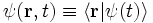
Since the position eigenstates form a basis for the state space, the integral over all projection operators is the identity operator:
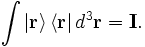
This statement is called the resolution of the identity. With this, and the fact that kets have unit norm, we can show that
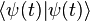 |
 |
 |
|
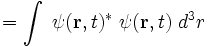 |
|
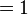 |
where ψ(r, t)* denotes the complex conjugate of ψ(r, t). This important result tells us that the absolute square of the wavefunction, integrated over all space, must be equal to 1:

We can thus interpret the absolute square of the wavefunction as the probability density for the particle to be found at each point in space. In other words, |ψ(r, t)|² d³r is the probability, at time t, of finding the particle in the infinitesimal region of volume d³r surrounding the position r.
We have previously shown that energy eigenstates vary only by a complex phase as time progresses. Therefore, the absolute square of their wavefunctions do not change with time. Energy eigenstates thus correspond to static probability distributions.
Operators in the position basis
Any operator A acting on the wavefunction is defined in the position basis by
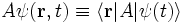
The operators A on the two sides of the equation are different things: the one on the right acts on kets, whereas the one of the left acts on scalar fields. It is common to use the same symbols to denote operators acting on kets and their projections onto a basis. Usually, the kind of operator to which one is referring is apparent from the context, but this is a possible source of confusion.
Using the position-basis notation, the Schrödinger equation can be written in the position basis as:
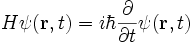
This form of the Schrödinger equation is the Schrödinger wave equation. It may appear that this is an ordinary differential equation, but in fact the Hamiltonian operator typically includes partial derivatives with respect to the position variable r. This usually leaves us with a difficult linear partial differential equation to solve.
Non-relativistic Schrödinger wave equation
In non-relativistic quantum mechanics, the Hamiltonian of a particle can be expressed as the sum of two operators, one corresponding to kinetic energy and the other to potential energy. For a single particle of mass m with no electric charge and no spin, the kinetic energy operator is
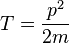
where p is the momentum operator, defined as

The potential energy operator is
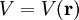
where V is a real scalar function of the position operator r. Putting these together, we obtain
![H \psi(\mathbf{r}, t) = (T + V) \, \psi(\mathbf{r}, t) = \left[ - \frac{\hbar^2}{2m} \nabla^2 + V(\mathbf{r}) \right] \psi(\mathbf{r}, t) = i \hbar \frac{\partial \psi}{\partial t} (\mathbf{r}, t)](http://en.wikipedia.org/math/f/4/b/f4b848c4d6276d6d94123dcad6b7ea45.png)
where 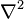 is the Laplace operator. This is a commonly encountered form of the Schrödinger wave equation, though not the most general one.
is the Laplace operator. This is a commonly encountered form of the Schrödinger wave equation, though not the most general one.
The corresponding time-independent equation is
![\left[ - \frac{\hbar^2}{2m} \nabla^2 + V(\mathbf{r}) \right] \psi(\mathbf{r}) = E \psi (\mathbf{r})](http://en.wikipedia.org/math/f/e/4/fe4dbc01201f1c512f436df93a26f308.png)
The relativistic generalisations of this wave equation are the Dirac equation, Klein-Gordon equation, Proca equation, Maxwell equations etc, depending on spin and mass of the particle. See relativistic wave equations for details.
Probability currents
In order to describe how probability density changes with time, it is acceptable to define probability current or probability flux. The probability flux represents a flowing of probability across space.
For example, consider a Gaussian probability curve centered around x0, imagine that x0 moving in a speed v toward the right. Then one may say that the probability is flowing toward right, i.e., there is a probability flux directed to the right.
The probability flux j is defined as:
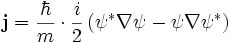
and measured in units of (probability)/(area × time) = r−2t−1.
The probability flux satisfies a quantum continuity equation, i.e.:
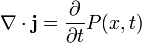
where P(x, t) is the probability density and measured in units of (probability)/(volume) = r−3. This equation is the mathematical equivalent of probability conservation law.
It is easy to show that for a plane wave,
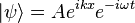
the probability flux is given by
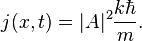
Solutions of the Schrödinger equation
Analytical solutions of the time-independent Schrödinger equation can be obtained for a variety of relatively simple conditions. These solutions provide insight into the nature of quantum phenomena and sometimes provide a reasonable approximation of the behavior of more complex systems (e.g., in statistical mechanics, molecular vibrations are often approximated as harmonic oscillators). Several of the more common analytical solutions include:
- The free particle
- The particle in a box
- The particle in a ring
- The particle in a spherically symmetric potential
- The quantum harmonic oscillator
- The hydrogen atom or hydrogen-like atom
- The ring wave guide
- The particle in a one-dimensional lattice (periodic potential)
For many systems, however, there is no analytic solution to the Schrödinger equation. In these cases, one must resort to approximate solutions:
- Perturbation theory
- The variational principle underpins many approximate methods (like the popular Hartree-Fock method which is the basis of the post Hartree-Fock methods)
- Quantum Monte Carlo methods
- Density functional theory
References
- Griffiths, David J. (2004) Introduction to Quantum Mechanics (2nd ed.), Prentice Hall. ISBN 013805326X
- E. Schrödinger, Phys. Rev. 28 1049 (1926)
External links
- Linear Schrödinger Equation at EqWorld: The World of Mathematical Equations.
- Nonlinear Schrödinger Equation at EqWorld: The World of Mathematical Equations.

